G2C::Software
GeneNomenclatureUtils: Tools for annotating genes and comparing gene lists with community resources.
by Mike DR Croning and Seth GN Grant
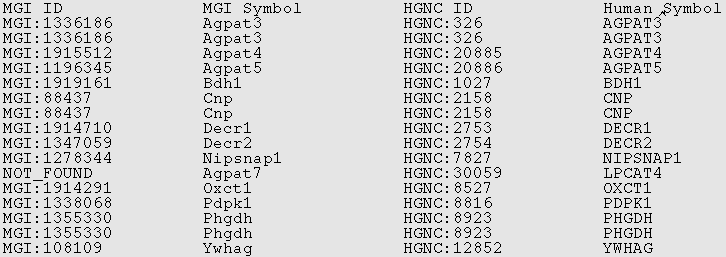
Example ids
Software Package and Collaborative Development
GeneNomenclatureUtils is hosted @ github
Introduction
Verifying, annotating, storing, and comparing lists of genes is now an essential task in modern experimental and computational biology. These lists can be derived from so-called 'omics' technologies that allow one to investigate gene (transcriptomics) or protein expression (proteomics) across the genome under defined conditions such as drug treatment, developmental stage, or clinical status. Similarly lists of candidate disease genes are being generated from the huge quantities of sequence data being produced by geneticists and clinicians employing new-sequencing technologies to identify sequence variation in large cohorts of human patient samples.
Comparing lists of genes produced by different labs from these disparate experimental sources remains an arduous, but obligatory task for the bioinformatician. This is because gene and protein identifiers are not stable, as they are continually revised and withdrawn over time by the gene nomenclature committees (such as HGNC) and database identifiers provided by other community genomic resources do also change. This 'identifier creep' is confounded by the fact that gene list comparisons often have to be made across species and model organism.
Here we provide an extensive suite of 40+ command-line driven utilities that are designed to simply this process. We provide an automated means to fetch the key nomenclature and other genomic, bibliographic and disease annotation resources from: HGNC, MGI, NCBI Entrez, OMIM, PubMed and UniProt, and store them on local disk. Required MEDLINE records are cached in a local MySQL database. This ensures a high-availability of all the resources, and ensures a consistent set of nomenclature and annotation can be used across the lifetime of an ongoing experiment or analysis.
GeneNomenclatureUtils is free software, collaboratively-developed, easy to install, and should prove useful to both bioinformaticians and experimental investigators, working with lists of genes.
Open Source License
The GeneNomenclatureUtils package is made available under the terms of the Artistic License 2.0.
Read the full license and disclaimer.
Documentation
Tool categories
Nomenclature file download
Use this script to automatically download all the nomenclature and other bioinformatic database files utilised by the GeneNomenclatureUtils package.
- download_nomenclature_files
Gene and protein nomenclature utilities
These scripts use the Entrez, HGNC, MGI, and UniProt resources to do identifier checking, by going from gene symbol to gene ID (or vice versa), or to fetch othologous gene IDs when going from mouse <-> human genomes.
Approved names and synonyms can be added to files of gene symbols or IDs.
Protein sequences and reported mouse alleles can be fetched for mouse genes.
- Entrez Gene: http://www.ncbi.nlm.nih.gov/gene
- check_entrez_gene_ids
- extract_entrez_gene_info_by_tax_id
- extract_from_ncbi_gene2_accession
- HGNC: http://www.genenames.org
- add_hgnc_id_by_hgnc_symbol
- MGI: http://www.informatics.jax.org
- add_approved_name_by_mgi_id
- add_mgi_id_by_hgnc_id
- add_mgi_id_by_mgi_symbol
- add_mgi_symbol_by_mgi_id
- add_ortho_gene_id_by_mgi_id
- add_uniprot_acc_by_mgi_id
- add_synonyms_by_mgi_id
- combine_by_mgi_id_column
- generate_interpro_report_from_mgi_id
- get_mgi_alleles_by_mgi_id
- get_prot_seqs_by_mgi_id
- UniProt: http://www.uniprot.org
- check_uniprot_ids
- parse_uniprot_accs
List comparison
Allows an arbitrary number of lists of genes or symbols to be compared eaily using a configuation file system so that 10s or 100s of lists can be managed easily.
- list_comparator
MEDLINE cache
A system to locally cache MEDLINE records, automatically fetched from PubMed, using a MySQL database to both coordinate the requests for the MEDLINE records, and subsequently supply them.
A simple object-orientated interface to the database is provided to utilise the cache: package MedlineCacheDB;
- medline_cache_create_db_tables
- medline_cache_dump
- medline_cache_pubmed_fetcher
- medline_cache_requester
OMIM disease - http://www.omim.org
Used to add OMIM IDs and OMIM entry titles to a file of human genes specified by Entrez Gene ID. The various 'gene' and/or 'phenotype' records can be added.
- add_omim_by_entrez_gene_id
PubMed
Uses the orthologous gene identifiers assembled in a file to combine the synonyms and human mutation terms to generate a PubMed URL search, to look for reports of association to disease of human gene mutations
- generate_pubmed_disease_searches_from_gene_nomenclature_ids
TAB-delimited file manipulation
A comprehensive set of utilities to manipulate tab- delimited text files.
- add_row_numbers
- categorise_by_column
- compare_columns_output_differences
- count_column_occurrences
- count_distinct_values_in_column
- count_row_occurrences
- count_rows_and_columns
- dump_column_as_unique
- extract_from_file_by_id_column
- extract_from_file_by_column_numerical_range
- filter_by_column
- pad_tabs_to_column_width
- rearrange_and_cut_columns
- remove_trailing_whitespace
- sort_file_by_column
- txt_file_format_sniffer
- write_spreadsheet
Command Line Parameters
Introduction
Most of the scripts in the GeneNomenclatureUtils package take one Unix-formatted tab-delimited text file (use txt_file_format_sniffer to check your txt file format) as input (specified by --file) and do something (look-up an equivalent ID, filter by, check, etc) the IDs or symbols specified in the column parameter(s) required by the individual scripts.
Columns are numbered from 1, that being the left-most column, and output is written to STDOUT by inserting a column in the lines of the file, as specified by --output_column, with existing columns being pushed to the right.
All of the scripts are documented with their particular mandatory and optional parameters. This is output automatically if an incorrect or insufficient set of parameters are specified, enter:
'./check_entrez_gene_id<cr>' to view the parameters of the script that checks Entrez Gene IDs, which will produce:
NAME - check_entrez_gene_ids
COMMAND LINE PARAMETERS
Required parameters
--file file to check
--entrez_gene_id_column column containing IDs to check (>=1)
--tax_id tax_id
--output_column column to output check results (>=1)
Optional parameters
--skip_title skip first (title) row
--help|h
DESCRIPTION
<SNIP>
An example command line is usually included in the documentation, that uses the demonstration data and configuration files included in the package.
The most common parameters
- --file
- The file to use, check, or process
- --skip_title
- Skip the first line (row) of the tab-delimited file, as it contains titles, rather than data
- --output_column
- Specify the output column, for the added data, existing columns are pushed rightword
- --tax_id
- Specify a taxonomy ID e.g. 9606 (human)
- --column
- Specify the column to check, or process
- --columns
- Specify a set of columns to check, or process e.g. --columns=1,2,3
- --value
- Specify a single value to find, count, or filter by
- --help|h
- Output the documentation
- --dir
- Specify directory to read or write from
- --file_list
- Specify a file to read file names from
- --config_file
- Specify configuration file. Details of the correct format are included in all scripts
- --mode
- Used to select between the behaviours of scripts with more than one running mode
- --quiet
- Produce less logging output
Gene nomenclature, id and symbol parameters
This parameter group is used to specify gene symbols and IDs from nomenclature committees and databases including MGI, HGNC, and Entrez Gene
- --mgi_id_column
- Specify column containing MGI IDs e.g. MGI:95819
- --hgnc_symbol_column
- Specify column containing HGNC symbols e.g. GRIN1
- --hgnc_id_column
- Specify column containing HGNC IDs e.g. HGNC:4584
- --mgi_symbol_column
- Specify column containing MGI IDs e.g. Grin1
- --entrez_gene_id_column
- Specify column containing Entrez Gene IDs e.g. 2902 (human)
All the above examples about are for the gene encding the NR1 subunit of the glutamate receptor, NMDA subtype
See:
- http://www.informatics.jax.org/searches/accession_report.cgi?id=MGI:95819
- http://www.gene.ucl.ac.uk/nomenclature/data/get_data.php?hgnc_id=HGNC:4584
- http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&...cmd=Retrieve&dopt=full_report&list_uids=2902
PubMed
- --pubmed_id_column
- Specify column containing PubMed IDs (PMIDs)
Other parameters
- --allow_dups
- Allow duplicates
- --ascending
- Sort or output in an asencding fashion (in string or numeric terms)
- --compare
- -
- --data_file
- data file from which to extract
- --data_file_id_column
- data file column with ids
- --descending
- Sort or output in a descending fashion (in string or numeric terms)
- --file1
- First tab-delimited data file
- --file1_column
- Column to check or process from file1
- --file2
- Second tab-delimited data file
- --file2_column
- Column to check or process from file2
- --filter_by
- -
- --has_title
- -
- --ids_file
- file holding IDs
- --ids_file_column
- column to use from IDs file
- --ignore_case
- Ignore case when sorting, processing a file
- --include_all
- -
- --lower_bound
- -
- --negative
- -
- --output_attrib
- -
- --output_ids
- -
- --output_mode
- -
- --output_unavailable
- -
- --positive
- -
- --suppress_title
- -
- --transfer_column
- Specify additional columns to copy from the input file to output
- --upper_bound
- -
- --width
- -
Testing and debugging parameters
- --test
- Switch on testing mode
- --debug
- Switch on debugging mode
Example Usage
- Install the package as described in: GeneNomenclatureUtils/docs/package_installation.txt
- Review the remaining documentation
- cd GeneNomenclatureUtils/scripts
Example 1 - add MGI IDs to the of mouse gene symbols, in the process checking the symbols
- Command
- ./add_mgi_id_by_mgi_symbol --file=../data/my_list_of_genes_1.txt --mgi_symbol_column=1 --output_column=2 --skip_title > ../example_output/example_1_output.txt
- Comment
- Note at least two 'NOT_FOUND' in the output file, for gene 'Agpat7', 'Lpaat-alpha'
Example 2 - add orthologous human HGNC IDs to the results of example 1
- Command
- ./add_ortho_gene_id_by_mgi_id --file=../example_output/example_1_output.txt --mgi_id_column=2 --output_column=3 --skip_title --output_attrib=human_hgnc_id > ../example_output/example_2_output.txt
- Comment
- Inspect the output file, a number of HGNC IDs have been added
Example 3 - add Human gene symbols to the results of example 2
- Command
- ./add_ortho_gene_id_by_mgi_id --file=../example_output/example_2_output.txt --mgi_id_column=2 --output_column=4 --skip_title --output_attrib=human_gene_symbol > ../example_output/example_3_output.txt
- Comment
- Inspect the output file, note mouse and human gene symbols are generally the same except human are capitalised
Example 4 - add approved names to the results of example 3
- Command
- ./add_approved_name_by_mgi_id --file=../example_output/example_3_output.txt --mgi_id_column=2 --output_column=5 --skip_title > ../example_output/example_4_output.txt
- Comment
- Inspect file, not names have been added
Example 5 - sort the results of example 4 by the added human gene symbols
- Command
- ./sort_file_by_column --file=../example_output/example_4_output.txt --column=4 --mode=string --ascending --skip_title > ../example_output/example_5_output.txt
- Comment
- Inspect file, note it has been sorted
Example 6 - Look for duplicates in the MGI IDs present in results of example 5.
- Command
- ./count_distinct_values_in_column --file=../example_output/example_5_output.txt --column=2
- Comment
-
Compare to output below:
./count_distinct_values_in_column ================================= Assumes a column title line Column number: 2 Column title : 'MGI ID' Distinct values: 13 Duplicates MGI:1336186 - 2 MGI:1355330 - 2 MGI:88437 - 2 NOT_FOUND - 2 Total number of duplicates: 4
Example 7 - Make a unique list of MGI IDs from the results of example 5
- Command
- ./dump_column_as_unique --file=../example_output/example_5_output.txt --column=2 --skip_title > ../example_output/example_7_output.txt
- Comment
- Inspect file, note it still contains a single 'NOT_FOUND'
Example 8 - Add orthologous human Entrez Gene IDs to the results of example 7
- Command
- ./add_ortho_gene_id_by_mgi_id --file=../example_output/example_7_output.txt --mgi_id_column=1 --output_column=2 --skip_title --output_attrib=human_entrez_gene_id
- Comment
- Inspect file, note the numerical IDs added
Example 9 - Using the result of Example 8 search OMIM for human disease phenotypes
- Command
- ./add_omim_by_entrez_gene_id --file=../example_output/example_8_output.txt --entrez_gene_id_column=2 --skip_title --mode=phenotitle --output_column=3 > ../example_output/example_9_output.txt
- Comment
- Note at least two of the genes have OMIM entries, for enzymatic deficiencies
Example 10 - Get synonyms for the genes specified by MGI in the results of example 7
- Command
- ./add_synonyms_by_mgi_id --file=../example_output/example_7_output.txt --mgi_id_column=1 --output_column=2 --skip_title > ../example_output/example_10_output.txt
- Comment
- Inspect the output file, note not all genes have synonyms
Example 11 - Check the txt file format of the first example file
- Command
- ./txt_file_format_sniffer --file=../data/my_list_of_genes_1.txt
- Comment
-
Depending how you obtained the package from Github the results could vary, example output below:
./txt_file_format_sniffer ========================= File : ../data/my_list_of_genes_1.txt Lines: 18 Unix/Linux - YES: Containing: \w\n\w DOS/Windows - NO : Containing: \w\r\n\w|\w\n\r\w MAC - NO : Containing: \w\r\w
Example 12 - Rearrange the order of the columns in the example 5 output, to place the MGI IDs first
- Command
- ./rearrange_and_cut_columns --file=../example_output/example_5_output.txt --columns=2,1,3,4,5 > ../example_output/example_12_output.txt
- Comment
- Inspect file, note rearrangement
Example 13 - Find the InterPro domains (and their frequency) in the mouse genes specified in the results example 12
- Command
- ./generate_interpro_report_from_mgi_id --file=../example_output/example_12_output.txt --mgi_id_column=1 --skip_title --output_mode=by_abundance > ../example_output/example_13_output.txt
- Comment
- Inspect the output file, note the most frequently occurring InterPro family/domain is IPR016040, NAD(P)-binding domain
Example 14 - Find mice alleles for the genes specified in the results of example 7
- Command
- ./get_mgi_alleles_by_mgi_id --file=../example_output/example_7_output.txt --mgi_id_column=1 --skip_title > ../example_output/example_14_output.txt
- Comment
- Notice from the STDERR output that (at least) 7 of the genes have reported alleles, and from the Pdpk1 has the most
Example 15 - Convert the results of example 14 into a Microsoft Excel format spreadsheet
- Command
- ./write_spreadsheet --file=../example_output/example_14_output.txt
- Comment
- Note from the output to screen that 14 rows were written to a file called example_14_output.xls
Example 16 - Generate URLS to search PubMed for human disease association of the genes from example 5
- Command
- ./generate_pubmed_disease_searches_from_gene_nomenclature_ids --file=../example_output/example_5_output.txt --mgi_id_column=2 --hgnc_id_column=3 --mode=disease --skip_title > ../example_output/example_16_output.txt
- Comment
- You may see warnings from the programme, these are non-fatal, examine the output file and cut and paste the search for Decr2 into http://www.ncbi.nlm.nih.gov/sites/entrez?db=PubMed
Example 17 - Setup the MySQL database for the MEDLINE cache
- Command
- ./medline_cache_create_db_tables --config_file=medline_cache_db.ini
- Comment
- Assuming you have created an empty database on your MySQL server, and set the connection parameters in GeneNomenclatureUtils/conf/medline_cache_db.ini, two tables will be created 'medline' and 'request'
Example 18 - Load some PubMed IDs requests into the MEDLINE cache db
- Command
- ./medline_cache_requester --config_file=medline_cache_db.ini --file=../data/my_list_of_pubmed_ids.txt --pubmed_id_column=1
- Comment
-
5 PubMed IDs should be parsed from the file:
./medline_cache_requester ========================= IDs parsed: 5 medline_entries_fetched : 0 pmids_already_available : 0 pmids_already_requested : 0 pmids_stored : 5 pmids_with_error : 0 total_pmids_parsed : 0
Example 19 - Fetch outstanding MEDLINE records
- Command
- ./medline_cache_pubmed_fetcher --config_file=medline_cache_db.ini
- Comment
-
If the fetch is successful you should get the following output:
./medline_cache_pubmed_fetcher ============================== Total entries to fetch: 5 URL Request size : 2 .. pmids_cached : 5 pmids_remaining : 0 pmids_requested : 5
Example 20 - Dump the fetched MEDLINE records to a text file
- Command
- ./medline_cache_dump --config_file=medline_cache_db.ini --file=../data/my_list_of_pubmed_ids.txt --pubmed_id_column=1 > ../example_output/example_20_output.txt
- Comment
- Inspect the output file it should contain 5 MEDLINE records
Example 21 - Compare two lists of gene identifiers
- Command
- ./list_comparator --config_file=example_list_config.yml --include_all > ../example_output/example_21_output.txt
- Comment
-
Inspect the output file, once should see two files were parsed and compared, and the comparision starts thus:
Comparison: MY-LIST-2 (11) UNION OF ALL SETS: 20 Gene MY-LIST-1 MY-LIST-2 MGI:1915512 YES NO MGI:1196345 YES NO MGI:1914291 YES NO MGI:107436 NO YES MGI:106672 NO YES MGI:108109 YES NO
Package Structure
The GeneNomenclatureUtils package has the following structure by default.
- GeneNomenclatureUtils
- GeneNomenclatureUtils/conf
- Example configuration files
- GeneNomenclatureUtils/data
- Example data files
- GeneNomenclatureUtils/docs
- Documentation
- GeneNomenclatureUtils/example_output
- Example output
- GeneNomenclatureUtils/modules
- Perl modules
- GeneNomenclatureUtils/scripts
- The package scripts
License
Copyright (C) 2005-2011 Genome Research Limited (GRL)
Authors: Mike Croning and Seth Grant
Refer to the Perl Artistic License 2.0 for the terms under which you may use modify, and redistribute this source. http://www.opensource.org/licenses/artistic-license-2.0.php
Comments and feedback to: mike_croning@hotmail.com
THIS PACKAGE IS PROVIDED "AS IS" AND WITHOUT ANY EXPRESS OR IMPLIED WARRANTIES, INCLUDING, WITHOUT LIMITATION, THE IMPLIED WARRANTIES OF MERCHANTIBILITY AND FITNESS FOR A PARTICULAR PURPOSE.